 |
Praveen Depa
|
 Email:
Email:
Duration in group: 2003 - 2007
Research Summary
Multiscale Simulation of Polymers
Introduction
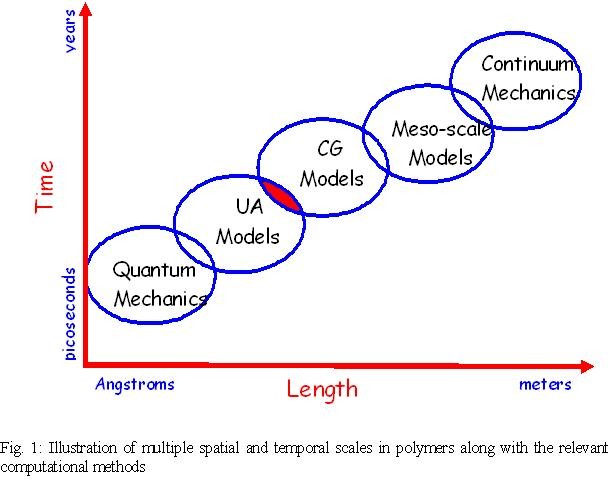
Many processes in polymeric materials occur during a large span of spatial and
temporal scales. Figure 1 shows various simulation techniques at the relevant
scales. In Molecular Dynamics [MD] Simulations, the positions of united atoms,
(a carbon along with its bonded hydrogens e.g. CH 4, CH3 etc.) are simulated
with force fields that are available in the literature. To reach larger length
and longer times, coarse-grained [CG] simulations are performed. CG simulations
are similar to MD, except that the united atoms are replaced by a collection of
united atoms called a coarse-grained bead. The problem with CG simulations is
the absence of accurate potentials or force fields to describe the interactions.
One solution to this is reverse mapping of the distributions from MD simulations
to obtain the force fields. The main objective of the research is to perform
simulations at atomistic and coarse-grained scales concurrently with the
exchange of information between them. This information is generally system
specific and examples of it are structure factors, energies etc.
Research objectives
My research involved the development of coarse-grained models
for a polyethylene melt. At present, four successive united atoms are grouped to
form a coarse-grained bead. Thus, every fifth united atom is considered as a
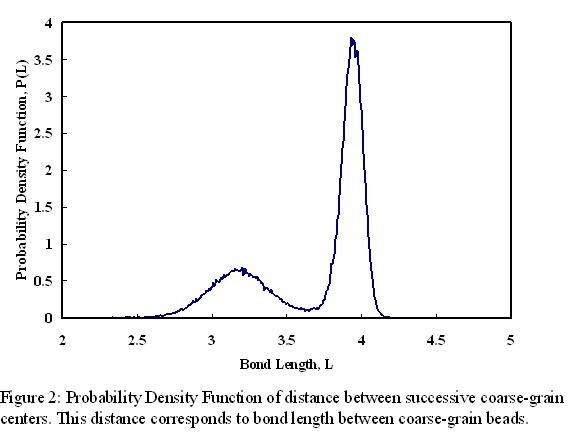
coarse-grained centre in a CG simulation. Figure 2 shows the probability
distribution function [PDF] of distance between successive coarse-grained
centers from the MD simulation. This corresponds to a coarse-grained bond length
in the CG simulation. CG bond length potential is then obtained by Boltzmann
inverting this PDF. Similarly, CG bond angle potential is obtained by Boltzmann
inverting the probability distribution of angle between 1-5-9 united atoms,
corresponding to a coarse-grained angle. Finally, inter-molecular interactions
are optimized to match the pair distribution function of coarse-grain centers
from the MD simulation. A CG simulation with these force fields gave static
properties that are comparable with those obtained from the MD simulation.
Simulations with higher degree of coarse graining
(mapping more than four united atoms on to a coarse-grain bead) are also
being performed. Future work involves extending this procedure to branched
polymers and polymer blends.
|
