Surface Science
Fundamental studies of interfacial phenomena are critical to many technologies. Over the years, our group has focused on understanding adsorption, surface diffusion, thermal desorption, assembly and interactions at surfaces, wetting, and interfacial free energies.
One recent application of interest to us is creating surfaces with controlled wettability. In particular, superhydrophobic surfaces have a number of beneficial properties, including water repellency, self-cleaning, low drag, and antifouling characteristics. Biological hydrophobic surfaces are characterized by structure over both micrometer and sub-micrometer length scales. This multi-scale functionality has inspired many studies aimed at synthesizing rough or patterned surfaces with superhydrophobic properties and quantifying the effect of roughness on superhydrophobicity. In our research, we employ continuum-level theory and atomic-scale MD simulations to establish the foundations for controlled wettability.
This work is supported by the US National Science Foundation.
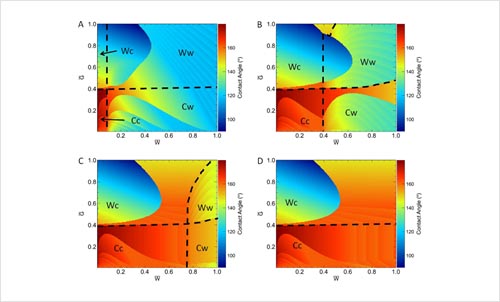
Effects of Hierarchical Surface Roughness on Droplet Contact Angle.
M. Bell, A. Shahraz, K. A. Fichthorn, and A. Borhan, Langmuir 31, 6752 (2015).
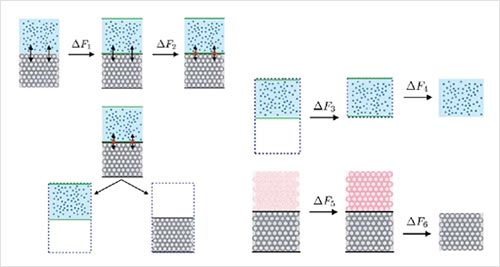
Obtaining the solid-liquid interfacial free energy via multi-scheme thermodynamic integration: Ag-ethylene glycol interfaces.
X. Qi, Y. Zhou, and K. A. Fichthorn, J. Chem. Phys. 145, 194108 (2016).
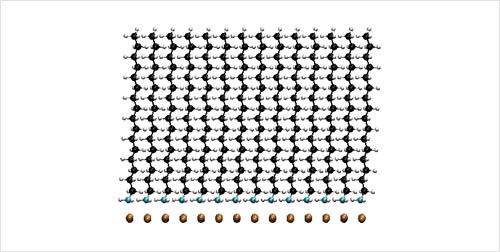
Self-Assembled Monolayer Structures of Hexadecylamine on Cu Surfaces: Density-Functional Theory.
S.-H. Liu, T. Balankura, and K. A. Fichthorn, Phys. Chem. Chem. Phys. 18, 32753 (2016).
Another topic of current interest is describing solid-liquid interfaces and their ramifications for the shapes of nanocrystals grown in solution. Key to achieving selective shapes are structure-directing agents (SDAs), or capping agents—solution-phase additives that promote the formation of specific nanocrystal shapes. It is widely held that SDAs promote nanocrystal shapes containing facets to which they bind the most strongly, but translating this preferential binding (predicted, for example, by first-principles calculations based on density-functional theory – DFT) into crystal shape requires accurate, multi-scale theory and simulations grounded in first principles. Our group is engaged in studying SDA adsorption from first principles, creating force fields based on DFT results, and conducting molecular-dynamics (MD) simulations based on these force fields to obtain information relevant for predicting nanocrystal shapes.
This work is supported by the US Department of Energy.